

O naší skupině
Vyvíjíme rychlé metody výpočetní chemie pro studium biomolekul. Kombinací semiempirických kvantově chemických výpočtů a technik strojového učení jsme schopni počítat velké systémy s tisíci atomů s nebývalou přesností. Tuto práci doplňují také přesné referenční výpočty a studium základních mechanismů nekovalentních interakcí.
Zaměřujeme se na výpočet vazebné afinity protein-ligand komplexů, což je klíčový krok v počítačovém návrhu léčiv. Naše metody kombinují jedinečné výhody a přesnost kvantově mechanických výpočtů s efektivitou potřebnou pro reálné aplikace.
Skupina spolupracuje s biochemiky a medicinálními chemiky v ÚOCHB a s předními farmaceutickými společnostmi na praktických aplikacích.
Publikace

SQM2.20: Semiempirical quantum-mechanical scoring function yields DFT-quality protein–ligand binding affinity predictions in minutes
Nature Communications 15: 1127 (2024)
Accurate estimation of protein–ligand binding affinity is the cornerstone of computer-aided drug design. We present a universal physics-based scoring function, named SQM2.20, addressing key terms of binding free energy using semiempirical quantum-mechanical computational methods. SQM2.20 incorporates the latest methodological advances while remaining computationally efficient even for systems with thousands of atoms. To validate it rigorously, we have compiled and made available the PL-REX benchmark dataset consisting of high-resolution crystal structures and reliable experimental affinities for ten diverse protein targets. Comparative assessments demonstrate that SQM2.20 outperforms other scoring methods and reaches a level of accuracy similar to much more expensive DFT calculations. In the PL-REX dataset, it achieves excellent correlation with experimental data (average R2 = 0.69) and exhibits consistent performance across all targets. In contrast to DFT, SQM2.20 provides affinity…
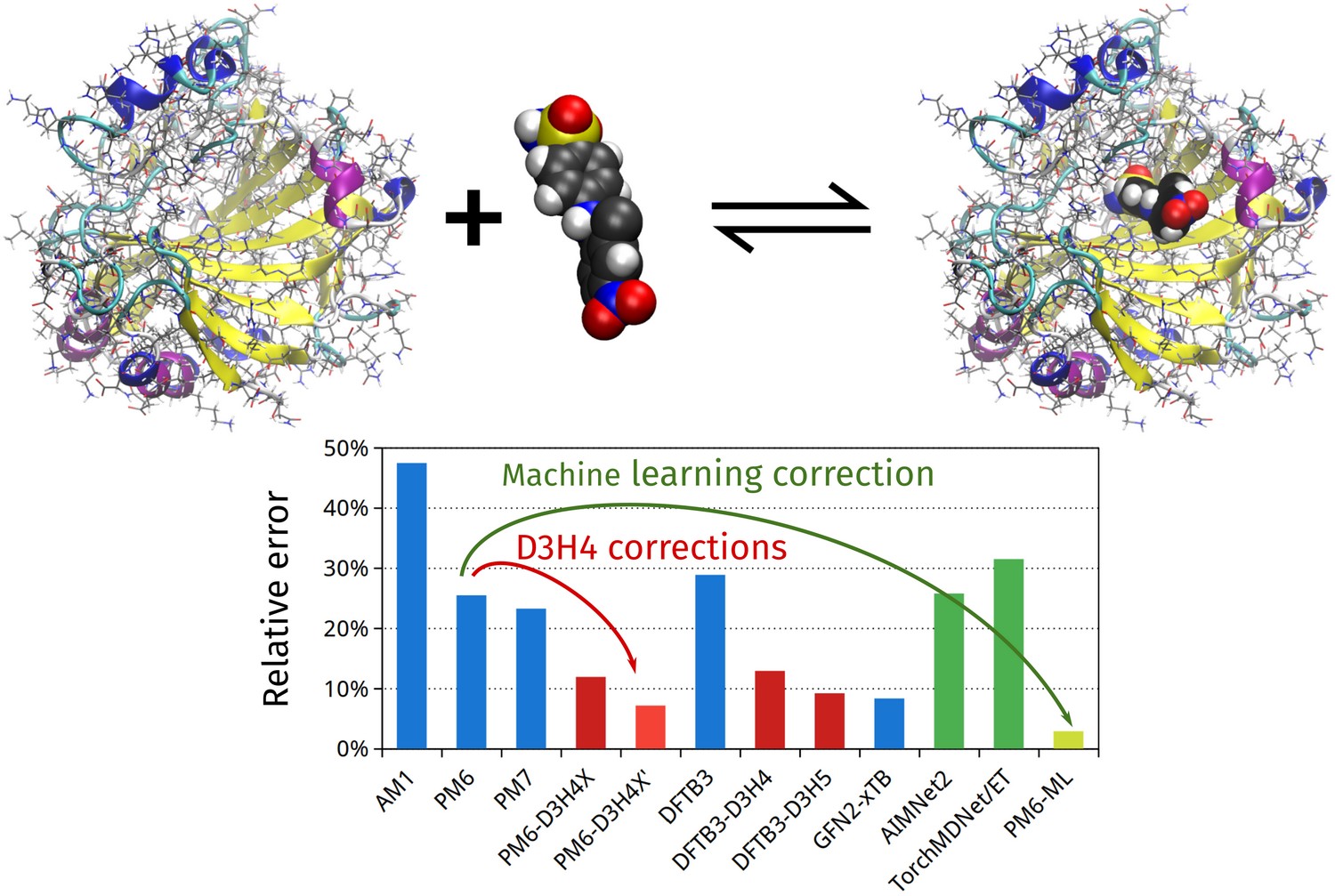
PM6-ML: The Synergy of Semiempirical Quantum Chemistry and Machine Learning Transformed into a Practical Computational Method
Journal of Chemical Theory and Computation 21 (2): 678–690 (2025)



